Most of the compounds we are investigating feature low lying (thermally accessible) excited states.
These excited states can couple with the ground state, and depending on their energy, can be thermally populated.
These small separations between states of different spin multiplicities allow reactions to cross from various potential energy surfaces to another, opening pathways to new types of reactivities.
However, this also complicates the assignment and correct interpretation of the ground state configuration (which may or may not interact with an excited state).
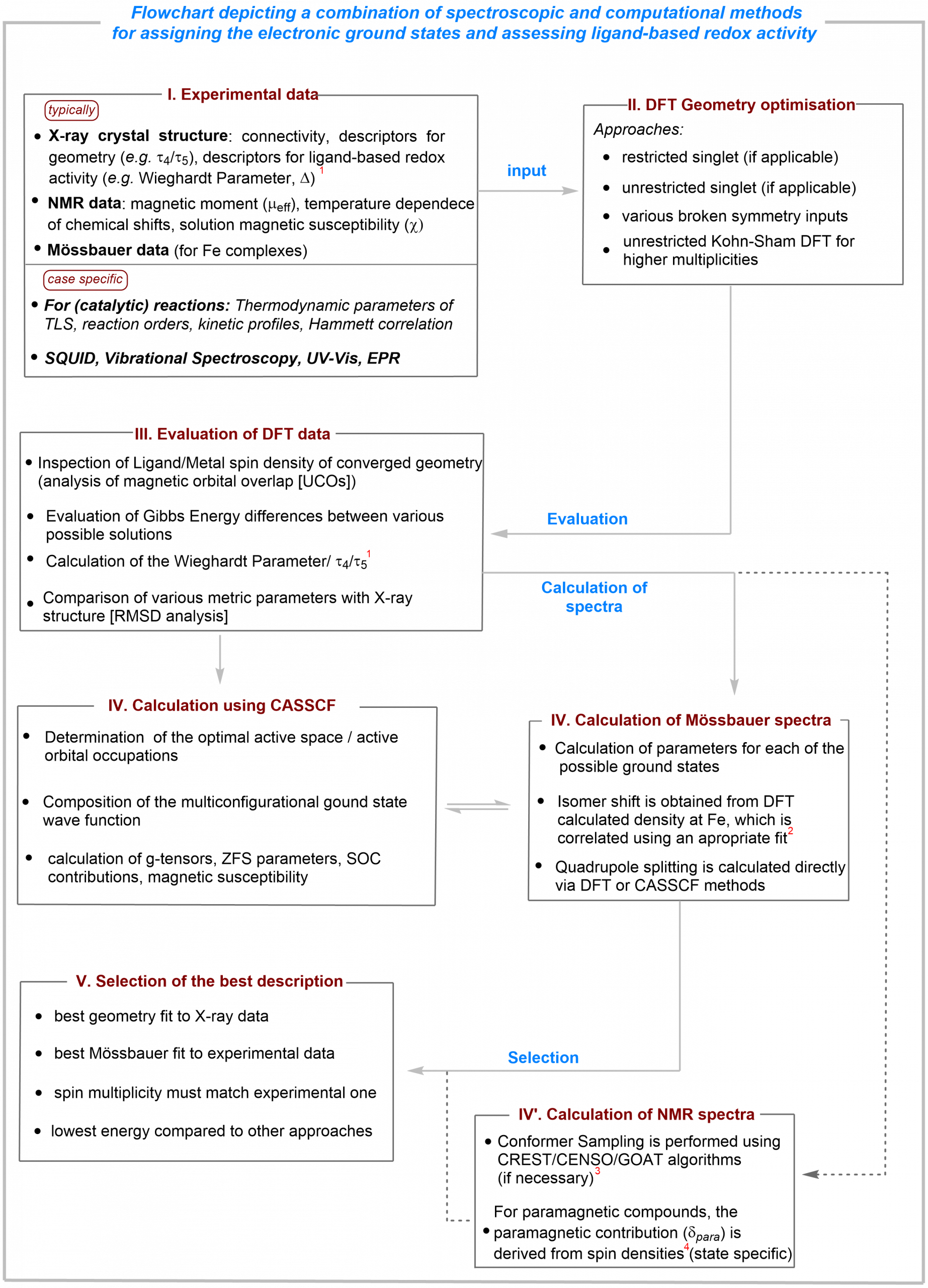
For this purpose, we typically use the following methodology to arrive at a more “realistic” description of the ground state:
(I) Where the stability of the compound allows it, experimental data are collected which usually includes X-ray structure, NMR spectroscopy and 57Fe Mössbauer spectroscopy (for iron-containing compounds).
These data already allow the extraction of the ground state magnetic properties and give hints on the iron oxidation state.
Variable temperature NMR studies allow us to determine if the magnetic ground state changes or is temperature dependent.
In some cases, SQUID magnetometry is necessary for a more accurate description.
(II) The connectivity model derived from X-ray diffraction, together with the ground state multiplicity (NMR spectroscopy) are used as input for DFT calculations.
To take into account ligand-based redox activity, the Broken Symmetry ansatz is used.
The energies of possible “pure” excited states are also calculated by varying the spin multiplicity.
(III) The data resulting from each ansatz are compared with the experimental data.
Special attention is given to how well a given functional reproduces the geometry obtained from experiment (X-ray diffraction) and the relative energies of the wavefunctions resulting from each approach.
(III’) In cases where the ground state clearly shows multiconfigurational character (which is in most cases in our present work), CASSCF calculations are performed.
The size of the active space is “controlled” by calculating the Mössbauer quadrupole splitting derived from each input and comparing it with the experimental one.
(IV) Using the DFT-derived geometry of each ansatz, the Mössbauer parameters (isomer shift and quadrupole splitting) are calculated.
These data are compared with the experimental ones.
(IV’) Most of the paramagnetic complexes in our work give well-resolved NMR spectra, which is due to the specific nuclear properties of the metal centre but also to the availability of low-lying excited states which accelerate electron-spin relaxation.
Given the large separation of 1H and 13C chemical shifts, in some cases, these can be calculated allowing the assignment of experimental data.
(V) The combination of the experimental and computational data furnishes, in most cases, a better educated guess of the likely description of the ground state.
{kind=link}